
Etablierung einer automatisierten Plattform zur Transkriptom- und Epigenomanalyse einzelner Zellen
Bei vielen Erkrankungen spielen epigenetische Modifikationen durch Methylierung von Cytosinen eine wichtige Rolle. Da sich Veränderungen des Epigenoms einer Zelle auf die Expression von Genen, also auf das Transkriptom der Zelle, auswirken, wäre es wünschenswert, epigenetische Veränderungen direkt mit der Expression auf RNA-Ebene korrelieren zu können. Im vorliegenden Projektantrag soll daher eine Methode zur gleichzeitigen Hochdurchsatz-Multiparameteranalyse von Epigenom und Transkriptom einzelner Zellen entwickelt und validiert werden. Die Methode soll dazu eingesetzt werden, Neurone und Mikroglia in Mausmodellen für ausgewählte neurodegenerative Erkrankungen auf Einzelzellebene zu charakterisieren. Im nächsten Schritt soll überprüft werden, ob sich entsprechende Signaturen auch in Blutzellen von erkrankten Tieren erkennen lassen. Die Methode soll parallel auch zu Charakterisierung von minimal-invasiv gewonnenen zirkulierenden Tumorzellen von Patienten mit Prostata-Karzinom verwendet werden.
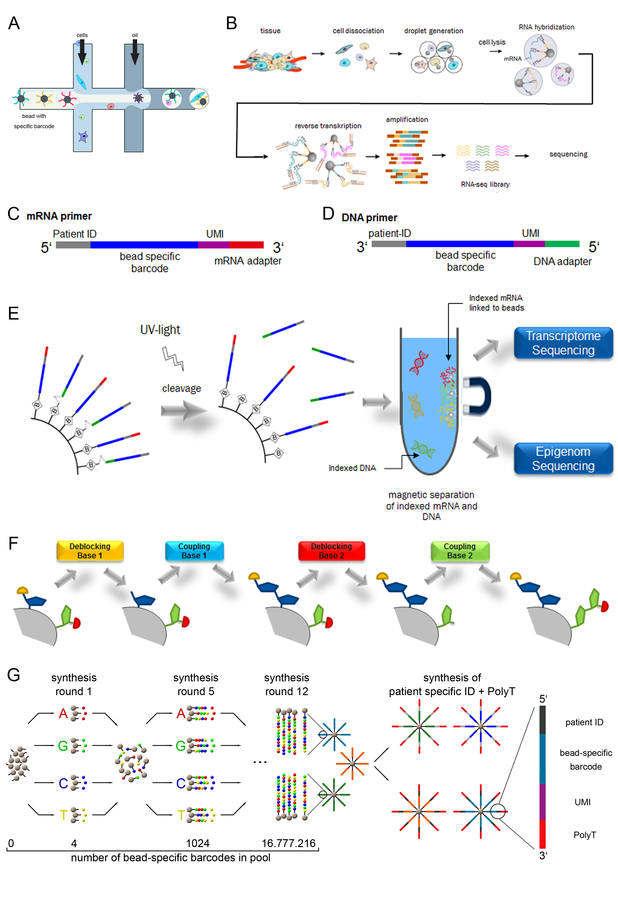
Parallele Sequenzierung von Trankriptom und Epigenom von individuellen Zellen im Hochdurchsatz-Verfahren. A, Schematische Darstellung des Prinzips des „Drop-Seq“ Verfahrens (modifiziert nach Macosko et al., 2015, Cell), Die Partikel, welche in Lysis-Reagenz suspendiert vorliegen, befinden sich im Zentralkanal, die Zellen treten über den oberen und unteren Kanal ein und werden mit dem Partikel in einem Tropfen eingeschlossen und dort lysiert. B, Heterogenes Gewebe wird zu Einzelzellen dissoziiert und im Droplet zusammen mit einem Mikropartikel mit individueller Primer-Sequenz eingeschlossen. Der „Barcode“ ist für alle Primer eines Mikropartikels gleich, aber einzigartig in Bezug auf die verschiedenen Mikropartikel. Nach Auflösung der Droplets können alle indizierten mRNA Fragmente amplifiziert und sequenziert werden. C, D, Aufbau des mRNA Primers (C) und DNA Primers (D), bestehend aus einer Patienten ID, dem Mikropartikel-spezifischem Barcode, der UMI (unique molecular identifier) und dem mRNA-Adapter (Poly-dT) beziehungsweise der DNA-Adapter-Sequenz. E, Schematische Darstellung zur Herstellung von Mikropartikeln, an die spaltbare DNA-Primer und nicht spaltbare mRNA-„Primer“ gekoppelt werden, die die gleiche spezifische Erkennungssequenz enthalten (siehe C und D). Nach Bindung der Primer an mRNA und DNA und der damit erfolgten Indizierung der Fragmente, kann die mRNA an den Mikropartikeln magnetisch von den DNA-Fragmenten separiert und beides unabhängig sequenziert werden. F, Die 2-fache Primersynthese wird über eine unabhängig kontrollierte Deblockierung und Kopplung von Basen an den mRNA- bzw. DNA-Primer realisiert, wodurch gleiche Sequenzen (Patienten ID und Mikropartikel-spezifischer Barcode) aber auch unterschiedliche Sequenzen (mRNA-Adapter bzw. DNA-Adapter) synthetisiert werden können. G, Schematische Darstellung des von Macosko et al. (2015) beschriebene „Split and Pool“ Verfahrens zur Generierung von spezifischen Erkennungssequenzen, die an Mikropartikel gekoppelt werden. Die Zuordnung der verschiedenen mRNA Spezies zu einer Zelle erfolgt nach Sequenzierung durch das Auslesen der zufällig generierten Erkennungssequenz.