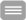
Methods
Next-Generation Sequencing (NGS)
In den letzten Jahren wurden neue Verfahren der Hochdurchsatz-Sequenzierung entwickelt, die unter dem Begriff Next-Generation Sequencing (NGS), zusammengefasst werden. Die Einführung der NGS-Technologien hat die Etablierung neuer diagnostischer Anwendungen in der täglichen Routine ermöglicht. So können die verschiedensten Krankheitsbilder bzw. Fragestellungen umfassend durch die gleichzeitige parallele Genanalyse bearbeitet werden, wodurch die klassische ‚Gen-für-Gen‘-Sequenzierung nach Sanger für komplexe Fragestellungen abgelöst wird. Im Vergleich mit der klassischen Sanger-Sequenzierung ermöglicht diese Technologie eine Steigerung des Durchsatzes bei gleichzeitig Kostenreduzierung.
Die einzelnen Schritte lassen sich im Wesentlichen in 4 Abschnitte gliedern: (A). Herstellung einer DNA-Bibliothek, (B). klonale, parallele Amplifikation der DNA, (C). die eigentliche Sequenzierungsreaktion und (D) Datenanalyse.

In unserem Labor besitzen wir eine MiSeq- und NextSeq-Plattform der Fa. Illumina, wodurch es uns möglich ist, eine große Anzahl an Proben parallel zu bearbeiten. Wir bieten verschiedene Multi-Gen-Panels, je nach klinischer Fragestellung, an (Link Diagnostik bzw. Begleitschein).
Anmerkungen:
Die Durchführung der Diagnostik sowie die Befunderhebung richtet sich nach der S1 Leitlinie: ‚Molekulargenetische Diagnostik mit Hochdurchsatzverfahren, beispielsweise mit Next-Generation- Sequencing‘ (https://www.gfhev.de/de/leitlinien/LL_und_Stellungnahmen/2017_09_15_GfH-S1-LL_NGS-Diagnostik_final.pdf). Das molekulargenetische Labor des Institutes für Humangenetik Jena nimmt regelmäßig an der vom Berufsverband Medizinische Genetik e.V. auf nationaler Ebene bzw. vom EMQN auf internationaler Ebene organisierten Qualitätskontrollen teil und ist nach DIN EN ISO 15189 akkreditiert.
- Aktuell ist die Abrechnung einer Paneldiagnostik mit den Krankenkassen (über den 10er Überweisungsschein), mit Ausnahme von Indikations-bezogenen EBM-Ziffern, nur bis zu 25 kb möglich (http://www.kbv.de/html/13259.php?srt=relevance&stp=fulltext&q=11513&s=Suchen; Stand 02/2018). Wird eine weiterführende Gen-Diagnostik gewünscht, muss ein schriftlicher Antrag bei der Krankenkasse gestellt werden ( Formulare....).
- Die Anreicherung erfolgt mittels Illumina TruSightTM Kits mit anschließender "paired-end" Sequenzierung.
- Analysiert werden alle kodierenden Exons sowie die Exon/Intron-Übergänge (+/- 10 bp).
- Zur Gewährleistung der diagnostischen Sicherheit wird eine Sequenziertiefe von mind. 20 Basen in 100% der analysierten Regionen gefordert.
- Der Befund enthält eine ausführliche Interpretation der Ergebnisse und der Methodenteil weist auf mögliche Einschränkungen hin.
- Potentiell pathogene Mutationen werden immer an einer 2. DNA-Isolierung bzw. 2. Blutprobe mittels Sangersequenzierung bestätigt.
- Limitierungen:
- Sequenzveränderungen im Promotor sowie bestimmte Rearrangements (z.B. Exon-übergreifende Duplikationen, Deletionen und Inversionen) werden mit dieser Analyse nicht sicher erfasst (die Analyse von Kopienzahlveränderungen erfolgt über MLPA, Array CHG).
- Das Vorliegen eines Zellmosaiks sowie balancierter Aberrationen kann durch diese Untersuchung nicht ausgeschlossen werden.
- Aufgrund der aktuellen Datenlage können Varianten im überwiegenden Teil der Intronbereiche, in den 5‘ bzw. 3‘ untranslatierten Bereichen sowie der Großteil synonymer Varianten hinsichtlich einer möglichen klinischen Konsequenz nicht oder nur bedingt beurteilt werden.
- Pathogene Mutationen in den nicht oder nur unzureichend abgedeckten Bereichen bzw. Bereichen mit niedriger Basenqualität sowie weiter intronisch gelegenen Mutationen können nicht ausgeschlossen werden.
- Repeat-Expansion von Genen sind nicht detektierbar.
- Die Analyse von Genen mit Pseudogenen ist aktuell nur bedingt möglich (z.B. CHEK2, PMS2).
Literatur: Yohe S und Thyagarajan B, Arch Pathol Lab Med. 2017 Nov;141(11); Koboldt DC et al., Cell 2013 Sept;155(1); http://www.annualreviews.org/doi/full/10.1146/annurev-genom-083115-022413; Behjati S und Tarpey PS, Arch Dis Child Educ Pract Ed. 2013 Dec;98(6)
Sanger sequenzierung
Die Didesoxymethode nach Sanger wird auch Kettenabbruch-Synthese genannt und stellt eine enzymatische Methode dar (von Sanger und Coulson um 1975 entwickelt). Bei dieser Methode werden neben der DNA-Polymerase und dem Nukleotid-Mix zusätzlich fluoreszenzmarkierte Stoppnukleotide (Dideoxy-Nukleotide) eingesetzt, bei deren Einbau es zu einem Abbruch der Reaktion an dieser Stelle kommt. Hierdurch entstehen fluoreszenzmarkierte Kettenabbruchprodukte unterschiedlicher Länge. Da die Stoppnukleotide mit 4 unterschiedlichen Fluoreszenzfarbstoffen markiert sind, kann man bei der Auswertung die einzelnen Basen unterscheiden und die Abfolge anhand der Größe der Fragmente bestimmen.

Die Methode findet im Wesentlichen Anwendung bei dem Verdacht auf das Vorliegen einer monogenen Erkrankung und bei der gezielten Mutationssuche.
In unserem Haus wird die Sangersequenzierung für die Gen-Diagnostik der HSP, HMSN/HNPP, von Zahnschmelzerkrankungen, dem Triple A-Syndrom und der hereditären Schmerzerkrankung (SCN9A), für die gezielte Mutationssuche sowie für die Bestätigungsdiagnostik von bekannten Mutationen angewandt (Verweis Begleitschein). Bekannte Pseudogenbereiche werden durch die Anwendung einer longrange-PCR analysiert.
- Limitierungen (s. auch NGS)
- Das Analyseergebnis kann durch SNPs (Single Nukleotide Polymorphism) in der Primerbindestelle beeinflusst werden.
- Das Vorliegen eines Zellmosaiks sowie balancierter Aberrationen kann durch diese Untersuchung nicht ausgeschlossen werden.
- Die Analyse von Genen mit Pseudogenen ist aktuell nur bedingt möglich (z.B. CHEK2, PMS2).
Literatur: http://journals.plos.org/plosone/article?id=10.1371/journal
Multiplex Ligation-dependent Probe Amplification (MLPA):
Diese Methode zur Detektion von Kopienzahlveränderungen (Deletionen/ Duplikationen) in nur einem Ansatz mit bis zu 50 verschiedenen DNA-Fragmenten wurde erstmals 2002 beschrieben (Schouten et al., Nucl Acids Res 30:12e57). Die MLPA-Reaktion erfolgt im Wesentlichen in vier Schritten: (1) DNA-Denaturierung und Hybridisierung der MLPA-Proben, (2) Ligationsreaktion, (3) PCR und (4) Trennung der Amplifikationsprodukte durch Kapillarelektrophorese. Kopienzahlveränderungen sind durch Vergrößerung bzw. Reduktion der Peakhöhen/-flächen im Vergleich zu den Kontrollen erkennbar. Jede der bis zu 50 MLPA-Proben in einem Ansatz besteht aus einer “Target”-spezifischen Sequenz, einer “Stuffer”-Sequenz definierter Länge und einer M13-Universal-Primersequenz. Die Multiplex-Amplifikation mit einem Fluoreszenz-markierten M13-Primerpaar kann nur nach Ligation der hybridisierten Proben erfolgen. Durch das Design der unterschiedlich langen “Stuffer”-Fragmente ist eine Trennung von Fragmenten einer Länge zwischen 130 und 480 bp möglich.
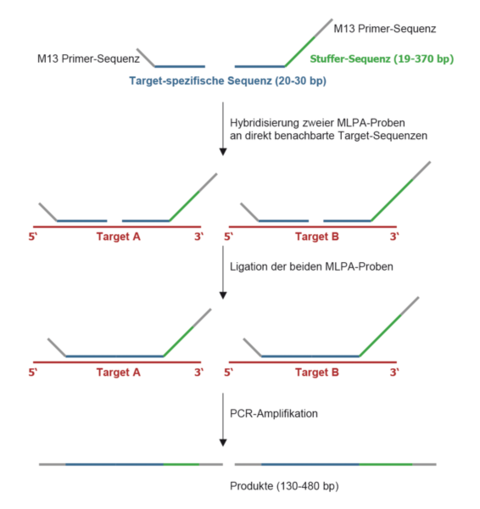
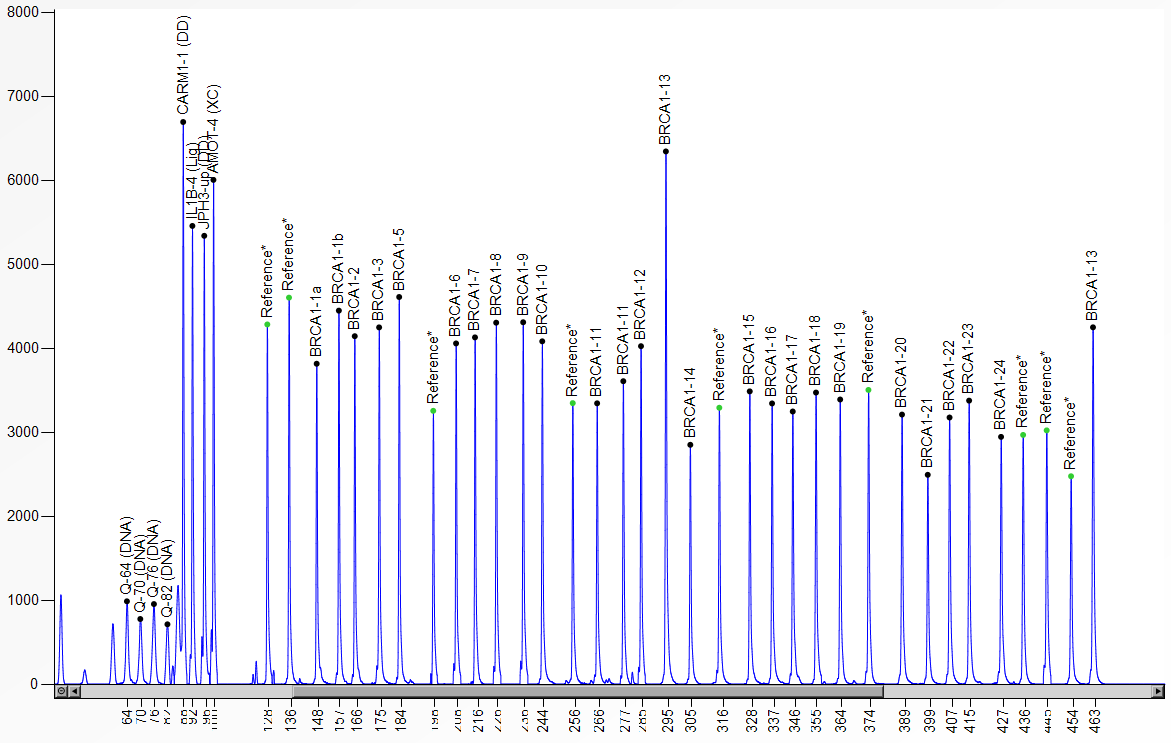
Coffalyzer-Auswerteprogramm, MRC Holland
Kommerziell verfügbar sind zahlreiche Kits zur Detektion von Kopienzahlveränderungen ganzer Gene, spezifischer Exons oder Chromosomenbereiche. Auch methylierungsspezifische MLPAs (MS-MLPA) zur gleichzeitigen Detektion von Kopienzahl- und Methylierungsveränderungen sind erhältlich. In unserem Haus kommen zahlreiche Kit’s für die unterschiedlichsten Fragestellungen zum Einsatz (s. Begleitschein mit *), wodurch vor allem auch in Kombination mit der NGS eine weitestgehend vollständige Genanalyse erfolgt.
- Limitierungen:
- Es kann nur eine Aussage über den Primer-abdeckenden bzw. –umspannenden Bereich in Abhängigkeit des verwendeten Kits getroffen werden.
- Das Analyseergebnis kann durch SNPs (Single Nukleotide Polymorphism) in der Primerbindestelle beeinflusst werden.
Literatur: https://www.mlpa.com
Array-CGH (Oligonukleotid-based)
Als weiterführende Methode zur Chromosomenanalyse zum Nachweis von Chromosomenveränderungen in Form von Deletionen/ Duplikationen kommt die Array-CGH (CGH: comperative genomic hybridisation) zum Einsatz. Die Array-CGH-Analyse erfasst das gesamte Genom, wobei die Detektionsrate von Kopienzahlveränderungen (CNV: copy number variation) von der Auflösung des verwendeten chip abhängig ist. Die aktuell für den diagnostischen Bereich geforderte Auflösung liegt bei min. 50 kb (http://www.kbv.de/html/13259.php?srt=relevance&stp= fulltext&q=11508&s=Suchen; Stand 02/2018). Das Prinzip beruht auf dem Einsatz von Oligonukleotiden, welche auf einer Matrix (chip) aufgebracht sind und repräsentativ das gesamte Genom abdecken. Die Oligonukleotide tragen Sequenzen, welche zum Genom komplementär vorliegen, so dass Patienten- und Referenz-DNA hybridisieren können. Aufgrund dieser vergleichenden Hybridisierung können sehr kleine submikroskopische unbalanciert vorliegende CNV bei den Patienten mittels software-Auswertung nachgewiesen werden. Zunehmend Anwendung findet der Einsatz polymorpher Oligonukleotide (‚SNP-Marker‘) in Kombination mit nicht-polymorphen Oligonukleotiden, wodurch der Nachweis von ‚Loss of Heterozygosity‘ ermöglicht wird (SNP-Array-Technik).
In unserem Labor kommt aktuell der 180K Oligonukleotid-chip mit einem mittleren Probenabstand von ca. 17 kb der Fa. Agilent zur Anwendung. Hierbei wird die Patienten- und Kontroll-DNA mit unterschiedlichen Fluoreszenzfarbstoffen markiert (Cy3/ Cy5) und zusammen hybridisiert. Mittels Laser-Scanner erfolgt die Detektion der beiden Fluorochrome und die nachfolgende Auswertung erfolgt software-basiert durch den Vergleich der beiden Fluorochromintensitäten.


- Limitierungen:
- Die Array-CGH-Methode ist limitiert auf die Detektion von Zugewinn bzw. Verlust an genomischem Material bis zur o.g. Auflösung.
- Punktmutationen, kleinere Deletionen, balancierte Translokationen und Inversionen, uniparentale Disomien (UPD), Imprinting-Defekte oder andere epigenetische Mutationen können nicht detektiert werden.
- Des Weiteren besteht die Möglichkeit, dass eventuell vorhandene Mosaike nicht erfasst werden.
- Allgemeine Informationen:
- Einer Array-CGH Analyse (an EDTA-Blut) muss eine Karyotypanalyse (an Heparinblut) vorausgehen zum Nachweis größerer struktureller Chromosomenaberrationen (http://www.kbv.de/html/13259.php?srt=relevance&stp= fulltext&q=11508&s=Suchen; Stand 02/2018).
- Die Kosten einer pränatalen Diagnostik werden bisher in Deutschland NICHT von den Krankenkassen übernommen (Stand 02/2018).
- Aktuell bieten wir keine Diagnostik von pränatalem Material an.
- Sollte sich nach erfolgter Stufendiagnostik (Karyotypanalyse und Array CGH), trotz bestehendem Verdacht, das Vorliegen eines Syndroms mit den genannten Methoden nicht bestätigen lassen, besteht die Möglichkeit einer weiterführenden NGS-Diagnostik (eine ausführliche klinische und familiäre Anamnese wird vorausgesetzt, Daten werden dem Labor zur Verfügung gestellt).
- Indikationen:
- Wiederholte Fehlgeburten in Eigen- oder Familienanamnese
- Subfertilität / Infertilität / vor Maßnahmen der künstlichen Befruchtung (ICSI/IVF)
- Verdacht auf strukturelle Chromosomenaberrationen, welche sich in der konventionellen Karyotypanalyse nicht bestätigt haben
- Verdacht auf ein Mikrodeletions- und Mikroduplikationssyndrom
- Multiple angeborene Fehlbildungen/ Dysmorphien
- Kleinwuchs/Großwuchs
- Mikrozephalie / Makrozephalie
- Autismus-Spektrum-Störung
- a. Familiarität einer bereits detektierten CNV
Literatur: Kharbanda M et al., Arch Dis Child Educ Pract Ed. 2015 Feb;100(1); Napoli E et al., J Autism Dev Disord. 2018 Feb;48(2); …
Microsatellite analysis
Nur ein geringer Teil (ca. 5 %) der menschlichen DNA codiert Gene für die Proteinsynthese, der weitaus größere Teil besteht aus Introns, regulatorischen Elementen und repetitiven DNA-Abschnitten, zu denen auch die Mikrosatelliten (STR = short tandem repeats) gehören.
- ‚Short tandem repeats‘ bestehen aus kurzen, sich wiederholenden Einheiten (Repeat) von 2-5 Basenpaaren, z.B.:
- GT GT GT GT GT usw. (Dinukleotidrepeat)
- GCT GCT GCT GCT (Trinukleotidrepeat)
- GTAG GTAG GTAG (Tetranukleotidrepeat)
Mikrosatelliten besitzen eine hohe Variabilität bezüglich der Anzahl der repeats. Da sie keinem Selektionsdruck unterliegen, sind sie i.d.R. nicht codierend und kommen verstreut im menschlichen Genom vor.
- Mikrosatellitenanalysen sind für verschiedene Fragestellungen einsetzbar, u.a.:
- LOH- (Loss of heterozygosity) Analyse
- Mikrosatelliteninstabilität (MSI)
- Bestimmung von krankheitsursächlichen repeat-Längen (HD, SCA)
- Nachweis von uniparentalen Disomien (UPD)
- Ausschluss/ Nachweis einer mütterlichen Kontamination
- pränatalen Schnelltest
- Herkunftsbestimmung von Marker-Chromosomen
- Gerichtsmedizin: Vaterschaftsnachweis, Täteridentifikation
Für die Untersuchung wird jeweils ein Primerpaar im Bereich der variablen Region gelegt und diese Region mittels PCR amplifiziert. Die Primer sind mit Fluoreszenzfarbstoffen markiert, so dass die entstehenden Produkte aufgetrennt und mittels Größenstandards die genaue Anzahl der Einheiten an einem Locus bestimmt werden kann. Die Auftrennung bzw. Auswertung erfolgt mittels Kapillarsequencer
- Limitierungen:
- Ein Mosaikanteil ist erst ab ca. ca. 10-15% sicher detektierbar.
- Die Bewertung kann nur für den Mikrosatelliten-abdeckenden Bereich erfolgen – somit können bspw. keine partiellen UPDs ausgeschlossen werden.
- Des Weiteren sind strukturelle Chromosomenveränderungen mit dieser Methode nur bedingt nachweisbar und bedarf bei Verdacht einer 2. unabhängigen Methode zur Bestätigung (Chromosomenanalyse, Array CGH, MLPA, FISH).
Literatur: Gymrek M, Curr Opin Genet Dev. 2017 Jun;44:9-16; Tang H et al., AJHG 2017 Nov;101:700-715