
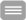
Hier finden Sie die aktuellen eigeninitiierten klinischen Studien, die von der Klinik für Innere Medizin II (Abt. Hämatologie/Onkologie) in Sponsorfunktion deutschlandweit durchgeführt werden.
Stand: Juni 2020
FAsciNation

Frontline Asciminib-Kombinationstherapie für CML-Patienten
Einschätzung der Wirksamkeit von Asciminib in Kombination mit Nilo-, Ima- und Dasatinib bei der Erreichung der tiefen molekularen Remission (MR4) in neudiagnostizierten CP CML‐Patienten im Rahmen einer prospektiven, nicht‐randomisierten, Phase II Kohortenstudie.
Aktuelles zu FAsciNation
04.09.2019 - Der erste Patient wurde in die FAsciNation-Studie eingeschlossen. Zur gemeinsamen Pressemitteilung der Universitätskliniken Jena und Leipzig

Studienkonzept
Geeignete Patienten mit de novo BCR-ABL positiver CML in chronischer Phase werden mit einem First-Line TKI nach Wahl des behandelnden Arztes anbehandelt. Dabei sind vor TKI-Behandlung maximal 4 Wochen Behandlung mit Hydroxyurea erlaubt.
Patienten, die für <6 Wochen mit Nilotinib 300 mg BID, Imatinib 400 mg QD oder Dasatinib 100 mg QD behandelt werden, sind geeignet für den Studieneinschluss und werden der jeweiligen Kohorte (je maximal 30 Patienten) zugeordnet.
Die Asciminib-Therapie wird 12 Wochen nach Beginn von Nilotinib, Imatinib oder Dasatinib und nach Wiederherstellung der normalen Hämatopoese begonnen.
Bei Intoleranz gegenüber der TKI-Initialtherapie vor Start von Asciminib sollten Studienpatienten die TKI-Therapie wechseln. Diese Patienten werden dann jedoch nicht mit Asciminib behandelt und verbleiben nicht in der Studie. Diese Patienten werden nicht in die Zählung der Kohorten aufgenommen.
Bei Wechsel des TKI unter Kombinationstherapie mit Asciminib wird die Asciminib-Therapie gestoppt und der Patient wird im Follow-Up bis zum Ende der Studie weiter beobachtet.
Nach 24 Monaten Therapie werden die Patienten auf der Grundlage ihres molekularen Ansprechens in Monat 24 drei Behandlungsarmen zugeteilt. Alle Patienten setzen die aktuelle Behandlung fort, bis die zugewiesene Therapie in Monat 25 begonnen wird.
- Patienten, die in Monat 24 keine MR4 erreicht haben, beenden Asciminib in Monat 25, verlassen die Studie und werden mit einem zugelassenen ATP-konkurrierenden TKI als Monotherapie nach dem Ermessen des Prüfarztes behandelt. Diese Patienten werden nur hinsichtlich des Überlebens weiterverfolgt.
- Patienten mit MR4 in Monat 24 setzen die bisherige Kombinationstherapie mit dem ATP-konkurrierenden TKI und Asciminib für ein weiteres Jahr (Monat 25 bis 36) fort.
- Bei Patienten mit MR4,5 oder besser in Monat 24 wird die Asciminib-Monotherapie mit 80 mg BID QD für ein weiteres Jahr (Monate 25 bis 36) verabreicht.
Nach Monat 36 wird die Asciminib-Behandlung für alle Patienten beendet. Die weitere Behandlung hängt von der Aufrechterhaltung einer tiefen molekularen Remission (MR4 oder besser) ab: Patienten mit MR4 oder besser für mindestens ein Jahr werden die Behandlung beenden und in eine TFR-Phase eintreten. Das PCR-Follow-up wird gemäß den aktuellen Empfehlungen durchgeführt. Patienten, die eine MR4 verloren haben, werden nach Ermessen des Prüfarztes mit dem ATP-konkurrierenden TKI weiterbehandelt und nur hinsichtlich des Überlebens weiterverfolgt.

Prüfzentren
Stand 04.11.2021
Eine Karte aller beteiligten Prüfzentren können Sie mit folgendem QR-Code
einsehen -->

Studienleitung und -koordination
Studiensponsor
Friedrich-Schiller-Universität Jena
Studienleiter
PD Dr. Thomas Ernst
Studienkoordinator und Ansprechpartner
Dr. rer. medic. Christian Fabisch
T +49 (0)3641 9-39 66 70
F +49 (0)3641 9-39 99 67
E
AsciNation bei ClinicalTrials.gov
Die folgenden Links werden nach Aktualisierung der Studienregister aktiviert:
DasaHit
Behandlungsoptimierung für Patienten mit chronischer myeolischer Leukämie (CML) mit Behandlung der natürlichen Krankheit (1. Linie) und Patienten mit Resistenz oder Intoleranz gegenüber alternativen Abl-Kinase-Hemmern (≥2. Linie)
Dasatinib Holiday for Improved Tolerability
In dieser klinischen Prüfung der Phase III wird untersucht, ob die Therapie mit Dasatinib an nur 5 Tagen in der Woche mit einer Wochenendpause gegenüber der herkömmlichen Dauertherapie mit Dasatinib (tägliche Medikation) bei gleicher Wirksamkeit besser verträglich ist.
Prüfdesign
Multizentrische, randomisierte, prospektive und kontrollierte Studie.

Insgesamt sollen ca. 300 erwachsene Patienten mit neu diagnostizierter Ph+ CML in der chronischen Phase oder Patienten mit Resistenz oder Intoleranz gegenüber vorausgegangener Therapie eingeschlossen werden. Eine Behandlung vor Studienbeginn mit Dasatinib oder Imatinib für max. 4 Wochen und mit Hydroxyurea für max. 6 Monate ist erlaubt.
Nach der Randomisierung (Aufteilen nach dem Zufallsprinzip) erhalten die Patienten 24 Monate entweder die Dauertherapie (Standard) oder die Therapie mit einer Wochenendpause (5 von 7 Tagen) Dasatinib. Während der Studienvisiten, die mindestens aller 3 Monate stattfinden, wird das Therapieansprechen und dessen Beständigkeit überprüft. Außerdem werden ein Nebenwirkungsprofil und die Lebensqualität anhand von Fragebögen beurteilt.
KONTAKT
Studienleiter
Wissenschaftliche und medizinische Anfragen
Prof. Dr. A. Hochhaus
Tel.: +49 (0) 3641 / 932 4201
Fax: +49 (0) 3641 / 932 4202
Medizinische Anfragen
Dr. T. Schenk
Tel.: +49 (0) 3641 / 932 4506
Fax: +49 (0) 3641 / 939 9967
Organisatorische Anfragen
Universitätsklinikum Jena
Klinik für Innere Medizin II
Abteilung Hämatologie und Internistische Onkologie
CML-Studienzentrale
Erlanger Allee 101, 07747 Jena
Tel.: +49 (0) 3641 / 939 6661
Fax: +49 (0) 3641 / 939 9986
E-Mail:
Datenmanagement/eCRF
ZKS Jena
Salvador-Allende-Platz 27
07747 Jena
Tel.: +49 (0) 3641 / 939 6653 o. -6690 o. -6677
Fax: +49 (0) 3641 / 939 9984
E-Mail:
Synopse (bitte anklicken)
Flyer zu der Studie (bitte anklicken)
Tiger Studie
Bis Juli 2017 wurden 717 erwachsene männliche und weibliche Patienten mit Ph und/oder BCR-ABL positiver CML in der chronischen Phase (CML-CP) in diese klinische Prüfung eingeschlossen. Die Patienten erhalten über einen Zeitraum von mindestens 24 Monaten eine sogenannte Induktionstherapie mit Nilotinib oder Nilotinib plus PEG-Interferon alpha 2b, an der sich wiederum für mindestens 24 Monate eine Erhaltungstherapie anschließt. Danach folgt eine behandlungsfreie Zeit, in der die Stabilität der molekularen Remission bis über 4 Jahre fortlaufend kontrolliert und im Falle eines molekularen Rückfalls die Therapie wieder aufgenommen wird. Die Hauptfrage der Studie ist, ob durch die Aktivierung des Immunsystems die Stabilität der Remission nach Absetzen der Therapie verbessert werden kann.
CML V
Tasigna and Interferon alpha evaluation initiated by the German CML Study Group
Ziel dieser klinischen Prüfung der Phase III ist die Untersuchung (i) des Effektes einer Kombinationstherapie mit Nilotinib und pegyliertem Interferon alpha im Vergleich zu Nilotinib allein auf die Rate guter molekularer Remissionen, (ii) der Stabilität der molekularen Remission unter PEG-Interferon Erhaltungstherapie und (iii) des Effektes der vorherigen PEG-Interferon-Therapie auf die Remissionsstabilität nach Absetzen aller Therapie.
Prüfdesign
Offene, multizentrische, randomisierte, Parallelgruppen-Studie mit neudiagnostizierten CML Patienten in der chronischen Phase.
Insgesamt sollen 652 erwachsene männliche und weibliche Patienten mit Ph und/oder BCR-ABL positiver CML in der chronischen Phase (CML-CP) in diese klinische Prüfung eingeschlossen werden. Die Patienten erhalten über einen Zeitraum von mindestens 24 Monaten eine sogenannte Induktionstherapie, an der sich wiederum für mindestens 24 Monate eine Erhaltungstherapie anschließt. Danach folgt eine behandlungsfreie Zeit über bis zu 4 Jahre, in der die Stabilität der molekularen Remission fortlaufend kontrolliert und im Falle eines molekularen Rückfalls die Therapie wieder aufgenommen wird.
Links/ weiterführende Informationen
Downloads
» Synopse
» Patientenseite mit Forum zu aktuellen Leukämie-News
Kooperationen
» SHG
» OSHO http://osho.uni-leipzig.de/
Ponderosa
Observational study on CML patients in any phase treated with ponatinib (Iclusig®) at any dose
Poster DGHO 2019
Rationale
Ponatinib ist seit Juli 2013 in der EU für Patienten, die resistent gegenüber anderen Tyrosinkinase-Inhibitoren sind, oder bei denen diese aufgrund von Unverträglichkeit nicht verabreicht werden können, zugelassen.
Neben der guten Wirksamkeit in der Drittlinientherapie eröffnet Ponatinib auch neue Möglichkeiten für Patienten, die aufgrund Ihres Mutationsprofils eine schlechte Prognose haben.
Aufgrund der bekannten Nebenwirkungen wird eine intensive Überwachung der behandelten Patienten von der European Medicine Agency (EMA) empfohlen.
In der Beobachtungsstudie sollen hierzu weitere Daten zur Behandlung mit Ponatinib in der täglichen Praxis gewonnen werden. Dazu gehören der Umfang und die Art von auftretenden unerwünschten Nebenwirkungen, ihrer Risikofaktoren und Behandlung sowie den medizinischen Folgen bei Patienten mit Chronischer Myeloischer Leukämie (CML) in allen Phasen der Erkrankung.
Primäre Ziele
- Ansprechen auf die Behandlung mit Ponatinib
- übliche Dosierung von Ponatinib im Behandlungsalltag
- Inzidenz von Unerwünschten Nebenwirkungen in der Studienpopulation
Sekundäre Ziele
- Progressionsfreies Überleben (PFS)
- Gesamtüberleben (OS)
Prüfdesign
- multizentrische prospektive und retrospektive Beobachtungsstudie
- mind. 100 Patienten in ca. 50 Prüfzentren
Indikation und wichtigste Einschlusskriterien
- Erwachsene Patienten mit CML in allen Phasen (CPCML, AP-CML, BP-CML) welche mit Ponatinib behandelt werden und ihre schriftliche Einwilligung für die Teilnahme an der Beobachtungsstudie gegeben haben.
- Patienten welche bereits nach Februar 2015 außerhalb klinischer Studien mit Ponatinib behandelt wurden können retrospektiv eingeschlossen werden.
Wichtigste Ausschlusskriterien
- Behandlung mit Ponatinib im Rahmen einer klinischen Studie nach AMG
- Behandlung mit weiterer Prüfmedikation im Rahmen einer klinischen Studie
- Behandlung der CML mit einem anderen TKI
- Stillende und/oder schwangere Patientinnen
Studienstart Juli 2015
Rekrutierungszeitraum bis ca. Dezember 2021
Studienende ca. 2023
Finanzielle Unterstützung durch:
Incyte Biosciences GmbH
Kontakt
Studienleiter, Wissenschaftliche und Medizinische Anfragen
Prof. Dr. A. Hochhaus
Tel.: +49 (0)3641 / 932 4201
Fax: +49 (0)3641 / 932 4202
Medizinische Anfragen
Dr. T. Schenk
Tel.: +49 (0)3641 / 932 4506
Fax: +49 (0)3641 / 939 9967
Organisatorische Anfragen
Universitätsklinikum Jena
Klinik für Innere Medizin II
Abteilung Hämatologie und Internistische
Onkologie
CML Studienzentrale
C. Clauß
Am Klinikum 1, 07740 Jena
Tel.: +49 (0)3641 / 939 6661
Fax: +49 (0)3641 / 939 9985
E-Mail:
Datenmanagement / eCRF
Tel.: +49 (0)3641 / 939 66 53
Fax: +49 (0)3641 / 939 99 84
Flyer zu der Studie (bitte anklicken)
BlastCrisis
Aktuelles zu BlastCrisis+
Rekrutierungsstart 08/2018
Studienkonzept
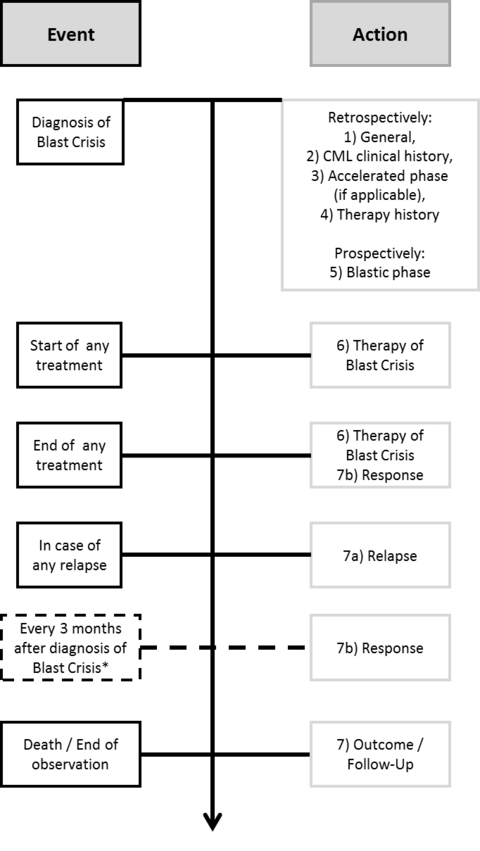
Die Blastenkrise in der Chronischen Phase der Myeloischen Leukämie (CML) ist ein seltenes Ereignis, welches den rapiden Krankheitsprogress mit einem Verhalten ähnlich einer akuten Leukämie darstellt. Die Lebenserwartung unbehandelter Patienten beträgt in dieser Phase nur noch wenige Monate, ist jedoch durch den Einsatz moderner Therapien in der vorherigen Chronischen Phase sehr selten geworden. Dennoch gibt es nur wenige Daten über das Eintreten, den Verlauf und die Behandlung von Patienten in dieser Phase der CML. Das Ziel des Registers ist daher diese Lücke zu schließen und aus den gewonnen Daten ggf. bereits erste gemeinsame Therapieempfehlungen oder Faktoren zu ermitteln, welche die Behandlung dieser Erkrankungsphase für Patienten und Ärzte verbessern können.
Im Register sollen zunächst 250 Patienten mit Blastenkrise in der CML erfasst werden. Es können sowohl prospektiv als auch retrospektiv Patienten mit Behandlung ab 01.05.2015 eingeschlossen werden. Es ist geplant das Register zunächst in Deutschland zu beginnen und im Verlauf weitere europäische Länder hinzuzuziehen um eine ausreichende Menge an Daten zu erhalten.
Studienlaufzeit
08/2018 bis 07/2021 – Rekrutierung
07/2018 bis 06/2024 – Datensammlung und Dokumentation
2024/2025 – Auswertung und Veröffentlichung
Studienleiter
PD Dr. Annamaria Brioli (Universitätsklinikum Greifswald)
PD Dr. rer biol. Hum. Michael Lauseker (IBE München)
Studienkoordinator und Ansprechpartner
Dr. rer. medic. Christian Fabisch
T +49 (0)3641 9-396670
M@il
OncoReVir

Register zur Erfassung von Virusinfektionen
der Atemwege
bei Patienten mit Krebserkrankungen
Auf dem Boden vor allem amerikanischer Studien und nur weniger deutscher Daten hat die AGIHO eine erste Leitlinie zur Diagnostik und Therapie von respiratorischen Virusinfektionen verfasst. Wünschenswert wäre für die Aktualisierung der Leitlinie und als wichtige Informationsgrundlage für behandelnde Onkologen eine deutliche Verbesserung der Datenlage zur Epidemiologie, zur Diagnostik und zu Behandlungsoptionen von respiratorischen Viren bei Krebspatienten in Deutschland.
Daraus resultieren folgende Fragestellungen: Welche CRV treten bei Patienten mit Krebserkrankungen vor allem auf? Welchen Verlauf nehmen CRV-Infektionen bei Patienten mit Krebserkrankungen, innerhalb und außerhalb des Settings der allogenen Stammzelltransplantation? Welche Patienten, insbesondere außerhalb des Settings der allogenen Stammzelltransplantation, sind besonders gefährdet für einen schweren Verlauf? Welche Rolle spielen die verschiedenen diagnostischen Verfahren? Sind die gängigen spezifischen Therapiemaßnahmen sinnvoll? Welchen Einfluss haben präventive Maßnahmen wie Impfungen und Hygienevorgaben?
Studienkonzept
multizentrisches, prospektives und retrospektives Register zur Erfassung von CRV-Infektionen bei Krebspatienten in Deutschland
Es sollen 1200-2000 Patientenfälle deutschlandweit in ca. 13 Prüfzentren eingeschlossen werden.
Prüfzentren
Stand 22.07.2020
Die ersten Zentren wurden bereits initiiert. Weitere Details folgen demnächst.
Studienleitung und -koordination
Studiensponsor
Friedrich-Schiller-Universität Jena
Studienleiter und medizinische Anfragen
Prof. Dr. med. Maria von Lilienfeld-Toal
Tel.: +49 (0) 3641 / 9-32 42 10
E-Mail: Marie.von_Lilienfeld-Toal@med. uni-jena.de
ITP-Register

Studienkonzept
Die Immunthrombozytopenie (ITP) ist eine seltene hämatologische Erkrankung, die durch einen autoimmun vermittelten Mangel an Thrombozyten zu einem höheren Blutungsrisiko bzw. einer verlängerten Blutungsdauer führen kann.
Bei Erwachsenen liegt die Inzidenz der ITP zwischen 2 bis 4 Neuerkrankungen pro 100.000 Einwohner/Jahr bei einer Prävalenz von 9 bis 26 pro 100.000. Daten zur Versorgungsrealität und Qualität der Behandlung der ITP in Deutschland werden derzeit nicht systematisch erfasst. Um die klinische Versorgung von Patienten mit ITP im Sinne der Versorgungsforschung zu charakterisieren und zu untersuchen, wird die Etablierung eines klinischen ITP-Registers am Universitätsklinikum Jena und die Möglichkeit zur Sammlung von Bioproben für die Biobank Dresden angestrebt.
Dies soll dem Aufbau der strukturellen Voraussetzungen für eine enge Kooperation der Prüfzentren dienen, um die Versorgungsqualität der betroffenen Patienten zu optimieren und eine Basis für den Ausbau weiterführender Forschungsaktivitäten zu schaffen.
Studienlaufzeit
12/2021 bis 12/2027 – Die Verlängerung auf eine kontinuierliche Erfassung der Patienten ist geplant
Studienleitung
Dr. Thomas Stauch (Universitätsklinikum Jena)
Dr. Karolin Trautmann-Grill (Universitätsklinikum Carl Gustav Carus Dresden)
Ärztlicher Leiter in Jena:
Dr. Thomas Stauch
E-Mail:
Studienkoordination
Zentrum für Klinische Studien Jena
Salvador- Allende-Platz 27
07747 Jena
Dr. Anne Ruschel (Klinisches Monitoring/Projektmanagement)
E-Mail:
Links:
Patientenwebsite zu ITP: https://www.leben-mit-itp.de/immunthrombozytopenie
Onkopedia Leitlinie: https://www.onkopedia.com/de/onkopedia/guidelines/immunthrombozytopenie-itp/@@guideline/html/index.html
VARIANT
Venetoclax nach TKI-Therapie zur Eliminierung persistierender Stammzellen bei CML
Mit dieser Studie soll untersucht werden, ob eine vorübergehende Venetoclax-Erhaltungstherapie nach Absetzen eines TKI den Anteil der Patienten erhöht, die die TKI-Behandlung dauerhaft absetzen können. Dies würde es ermöglichen, chronische Nebenwirkungen, potenzielle Langzeittoxizitäten und erhebliche jährliche Kosten zu vermeiden. Eine dauerhafte Beendigung einer TKI-Therapie wäre somit von erheblichem Wert für die Patienten und die Gesellschaft.
Aktuelles zu VARIANT
15.06.2023 – Die Studie wurde von den Behörden genehmigt.

Studienkonzept
Zehn Patienten mit BCR::ABL1-positiver CML in chronischer Phase, die seit mindestens drei Jahren mit TKI behandelt werden und seit mindestens einem Jahr mindestens MR4 erreicht haben, werden nach Absetzen der TKI 12 Monate lang mit 400 mg Venetoclax täglich behandelt. Die molekulare Überwachung erfolgt durch eine international standardisierte qPCR für BCR::ABL1 mRNA-Transkripte und einen neuartigen hausinternen genomischen digitalen PCR-Assay zur Messung der verbleibenden CML-Stammzellen in tiefer/kompletter molekularer Remission. Der primäre Endpunkt ist die Reduktion der BCR::ABL1-Stammzellen im Knochenmark nach Venetoclax-Verabreichung mittels quantitativer genomischer PCR. Sekundäre Endpunkte sind das molekulare rückfallfreie Überleben (RFS), wobei ein Rückfall als Verlust der großen molekularen Remission (MMR) definiert ist, d.h. ein Anstieg des BCR::ABL1-Spiegels (IS) auf > 0,1%. Weitere sekundäre Endpunkte sind die Bewertung der Sicherheit der Venetoclax-Behandlung bei CML und die Lebensqualität vor und nach Absetzen des TKI.
Bei Intoleranz oder anderen Problemen unter Therapie mit Venetoclax wird die Therapie gestoppt und der Patient wird im Follow-Up bis zum Ende der Studie weiter beobachtet.
Prüfzentren
Universitätsklinikum Jena Apl. Prof. Dr. med. Thomas Ernst
Uniklinik der RWTH Aachen Dr. med. Martina Crysandt
Studienleitung und -koordination
Studiensponsor
Studienleiter
Studienkoordinator und Ansprechpartner
Dr. rer. medic. Christian Fabisch
T +49 (0)3641 9-39 66 70
F +49 (0)3641 9-39 99 73
E