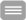
We are an international team of physicians, biologists, biochemists and related disciplines which share an interest for the pathophysiology of monogenic hereditary disorders. Main topics are currently ion transport related disorders, several of which resulting in altered receptor functions, and neurodegenerative disorders like axonopathies, some caused by mutations in genes encoding receptors. To unravel the chain of events that finally result in a specific pathology we use different approaches including molecular biology, cell models, the generation of mouse models using homologous recombination and CRISPR/Cas mediated genome engineering, new sequencing technologies, and advanced microscopy techniques.
Functional Genetics

Ion transport related disorders
Ion transport processes play crucial roles for neuronal excitability, signal transduction, transport across epithelia, and the homeostasis of extracellular, cytosolic, and vesicular compartments. The essential role of the molecules involved in these processes is reflected by a large number of hereditary disorders due to mutations in genes encoding ion channels or -transporters. Over the past years our group has established and analyzed several knockout-mouse models of ion channels/ -transporters to better understand their pathophysiological role in neurological and kidney related diseases.
![<b>Figure 1: GABA and Chloride homeostasis.</b> In immature neurons the intracellular Cl- concentration [Cl-]i is high because of NKCC1 mediated Cl- accumulation. Hence, opening of GABAA receptors results in an influx of Cl-. During development the incipient expression of KCC2 lowers [Cl-]I and renders GABA hyperpolarizing. The expression of KCl cotransporters can change in pathological conditions such as epilepsy, brain trauma or chronic pain, thus modulating GABAergic transmission.](/humangenetik_media/Dokumente/Arbeitsbereiche/Funktionelle+Genetik/Fig1_+GABA+and+Chloride+homeostasis-font-931-p-1384.png)
Many neurotransmitter receptors are ligand-gated ion channels: Upon binding of the respective ligand a conformational change opens the ion channel, which leads to a flow of ions across the cell membrane. This, in turn, results in either a depolarization, for an excitatory receptor response, or a hyperpolarization, for an inhibitory response. Neurotransmitter is released into the synaptic cleft by fusion of presynaptic vesicles with the presynaptic membrane. The neurotransmitters can then bind to receptors located on the postsynaptic neuron. Because the existing electrochemical gradients determine the ion flow, a tight control of ion homeostasis is crucial for brain function. We and others could show that the cation chloride co-transporters KCC2/SLC12A5 (Pubmed 11395011) and NKCC1/SLC12A2 (Pubmed 19295148) are crucial in the control of the electrochemical Cl- gradient that is required for “classical” hyperpolarizing postsynaptic inhibition mediated by GABAA and glycine receptors (Figure 1). Supporting this model mutations in the gene encoding KCC2 have been linke to monogenic epilepsy. We currently address the role of these and related transporters in the context of brain trauma within an international research group headed by Claudio Rivera (ACROBAT) and during cortical development (Priority program of the DFG).
Because GABAA receptors also conduct HCO3- we extended our studies on HCO3- transporters of the SLC4 family of genes such as the Na+ coupled anion-exchangers NDCBE/SLC4A8, which modulates excitatory transmission by modulating glutamate release (Pubmed 21593314. NCBE/SLC4A10 is closely related to NDCBE and also contributes to neuronal pH homeostasis and neuronal excitability (Pubmed 18165320). Moreover, SLC4A7 plays an essential role for the production of the cerebrospinal fluid by mediating basolateral Na+ entry into choroid plexus epithelial cells. Thus mice devoid of NCBE show collapsed brain ventricles.
Ion transport processes are also critical for the composition of the “milieu interne”, which is a result of the dynamic and controlled exchange of solute and water between body cells and our surrounding environment. The equilibrium requires that the amount of water and ions excreted into the urine exactly matches the daily intake or production of various substances. The kidney plays a central role for electrolyte and water homeostasis. In humans the glomeruli filter daily very large amounts (∼180 liters) of water and of ions (e.g., 25,000 mmol of Na+, 4,300 mmol of HCO3−, 720 mmol of K+). These quantities easily exceed those in the body. Accordingly, the renal tubule must absorb most substances filtered at the glomerulus to avoid their loss into urine. However, as indicated above, a small fraction of water and ions that exactly matches the daily input from diet and metabolism must also be excreted.

Net elimination of nonvolatile acid is mediated by type A-intercalated cells (A-ICs) in the connecting tubule and collecting duct (CD). To achieve proton (H+) secretion, A-ICs express V-type ATPase at the apical cell pole. Protons are generated within these cells by hydration of CO2, which is catalyzed by intracellular carbonic anhydrase type II (CAII). The resulting H2CO3 dissociates into H+ and HCO3-. Whereas H+ is secreted apically via the proton pump, HCO3- is extruded basolaterally by the chloride/bicarbonate exchanger AE1 (SLC4A1). This transport model is supported by hereditary defects, also known as distal renal tubular acidosis (dRTA), either caused by mutations of either the a4 or B1 subunit of the proton pump, of CAII, or of the anion-exchanger AE1, respectively. Recently, we generated a knockin mouse model for the most common missense mutation in AE1 in patients suffering from dominant distal renal tubular acidosis (Pubmed 27335120). Our results suggest that reduced basolateral anion-exchange activity inhibits trafficking and regulation of the V-type ATPase, thus compromising luminal H+ secretion and possibly lysosomal acidification. In the past we had already shown that the KCl-cotransporer KCC4 is important for chloride recycling at the basolateral membrane of A-ICs (Pubmed 11976689) and thus supports basolateral HCO3- transport via the anion-exchanger AE1 (Figure 2). With a knockout mouse model for the a4 subunit we also identified a previously unrecognized role of the a4 subunit in proximal tubule cells. We could show that a4 KO mice suffer not only from severe acidosis but also from proximal tubule dysfunction with defective endocytic trafficking, proteinuria, phosphaturia and accumulation of lysosomal material.
Our finding that NDCBE/SLC4A8-dependent thiazide-sensitive Na⁺ reabsorption occurs in the cortical collecting duct challenges the current model of how thiazides mediate their antihypertensive action and identifies a potentially new target for antihypertensive strategies (Pubmed 27151921,Pubmed 20389022). Moreover, basolateral NaCl exit from β-intercalated cells was independent of the Na+/K+-ATPase but critically relied on the presence of basolateral SLC4A9/AE4 (Pubmed 23610411).
Chloride transport by the renal tubule is critical for blood pressure (BP), acid-base, and potassium homeostasis. Chloride uptake from the urinary fluid is mediated by various apical transporters, whereas basolateral chloride exit is thought to be mediated by ClC-Ka/K1 and ClC-Kb/K2, two chloride channels from the ClC family, or by KCl cotransporters from the SLC12 gene family. Nevertheless, the localization and role of ClC-K channels is not fully resolved. Because inactivating mutations in ClC-Kb/K2 cause Bartter syndrome, a disease that mimics the effects of the loop diuretic furosemide, ClC-Kb/K2 is assumed to have a critical role in salt handling by the thick ascending limb. To dissect the role of this channel in detail, we generated a mouse model with a targeted disruption of the murine orthologue ClC-K2. Mutant mice develop a Bartter syndrome phenotype, characterized by renal salt loss, marked hypokalemia, and metabolic alkalosis. Patch-clamp analysis of tubules isolated from knockout mice suggest that ClC-K2 is the main basolateral chloride channel in the thick ascending limb and in the aldosterone-sensitive distal nephron (Pubmed 27335120).
Axonopathies

The axon is the long process of a neuron that carries efferent action potentials from the soma to the target cell (Figure 3). Because protein and lipid synthesis largely occur in the cell soma, anterograde transport is required to supply axons with the respective materials. Conversely, material intended for degradation has to be transported in a retrograde manner to the cell soma. The same logistic challenge applies to intracellular signals, both anterogradely and retrogradely. Thus, it is no surprise that neurons with long projections are particularly susceptible to impairment in cell processes such as trafficking, transport, energy utilization, and signaling and cytoskeletal organization. The term axonopathy refers to a group of neurodegenerative disorders that mainly manifests at long axons. Hereditary spastic paraplegias (HSPs) are defined by the degeneration of corticospinal tract axons connecting the cortical motoneuron with spinal cord motoneurons, resulting in a progressive spastic gait disorder. In hereditary motor and sensory neuropathies (CMT/HMSN) degeneration of efferent lower motoneuron axons and afferent nerve fibers cause muscle weakness and loss and sensory deficits. In hereditary sensory and autonomic neuropathies (HSAN) also autonomic fibers are involved. The latter are often characterized by the absence of pain sensation.
In the past we generated a couple of mouse models for different HSP subtypes. E.g. we could show that REEP1 (receptor expression enhancing protein 1), which is mutated in autosomal dominant SPG31, is an endoplasmic reticulum (ER) resident protein, which contributes to ER structure via its RETICULON domain (Pubmed 24051375). We also addressed the function of Spastizin (SPG15, Pubmed 24367272) and Spatacsin (SPG11, Pubmed 26284655) for motoneuron maintenance. Both protein associate with a recently identified adaptor protein complex, i.e. AP-5, and are thought to be involved in the recycling of lysosomes from autolysosomes, while the role of AP-5 is largely unknown.

Our efforts on axonopathies also include polyneuropathies. E.g. we studied the role the K-Cl cotransporter KCC3 for Andermann syndrome, which is characterized by epilepsy, agenesis of the corpus callosum, and polyneuropathy (Pubmed 14532115, Pubmed 16424367). Together with Ingo Kurth we were able to identify the role of voltage-gated sodium ion channel Nav1.9 in individuals with the congenital inability to experience pain (Pubmed 24036948 ).
Moreover, we could show that mutations in FAM134B cause hereditary sensory and autonomic neuropathy type 2 (HSAN2) (Pubmed 19838196). Together with the group of Ivan Dikic we were later able to show that FAM134B serves as an LC3 receptor and thus triggers the autophagy of endoplasmic reticulum (ER) membranes, a process also referred to as reticulophagy or ER-phagy (Figure 4) (Pubmed 26040720). Thus FAM134B was the first ER resident receptor that controls ER turnover and ER size. We currently try to address how this process is regulated and how it relates to the unfolded protein response (UPR).

Another receptor associated with HSAN is NTRK1, one of three major receptor tyrosine kinases involved in neurotrophin signaling. Upon NGF binding, NTRK1 is autophosphorylated resulting in the activation of downstream signaling pathways that support neurite outgrowth, survival of sympathetic ganglia and nociceptive sensory neurons. Mutations in NTRK1 cause hereditary sensory and autonomic neuropathy type four (HSAN-IV), also known as congenital insensitivity to pain with anhydrosis (CIPA, OMIM 256800). Together with Ingo Kurth we established a NTRK1 deficient cell line using CRISPR/Cas. This system allows studying axon outgrowth upon expression of disease related variants in response to neurotrophin (Figure 5) and thus enables us to characterize signaling of variant receptors.
Selected readings
SLC12
Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ (2001) Disruption of KCC2 reveals an essential role of K-Cl-cotransport already in early synaptic inhibition. Neuron 30:515-24
Boettger T*, Hübner CA*, Maier H, Rust M, Beck FX, Jentsch TJ (2002) Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter KCC4. Nature 416:874-78
Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Völkl H, Hübner CA, Jentsch TJ (2003) Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO Journal 22:5422-34
Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hübner C.A., Represa A, Ben-Ari Y, Khazipov R (2006) Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314:1788-92
Rust MB, Faulhaber J, Budack M, Pfeffer C, Maritzen T, Didié M, Beck FX, Boettger T, Schubert R, Ehmke H, Jentsch TJ, Hübner CA (2006) Neurogenic mechanisms contribute to hypertension in mice with disruption of the K-Cl-cotransporter KCC3. Circulation Research 98:549-56
Rust MB, Alper SL, Rudhard Y, Brugnara C, Trudel M, Jentsch TJ, Hübner CA (2007) Disruption of erythroid K-Cl co-transporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. Journal of Clinical Investigation 117:1708-17
Pfeffer C, Stein V, Keating D, Maier H, Rudhard Y, Hentschke M, Rune G, Jentsch TJ, Hübner CA (2009) GABA-dependent network activity contributes to early maturation of excitatory hippocampal synapses. Journal of Neuroscience 2:3419-30
Antoine MW, Hübner CA, Arezzo JC, Hébert JM (2013) A causative link between inner ear defects and long-term striatal dysfunction. Science 341(6150):1120-3
Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL, Harvey L, Sun D, Hübner CA, Cull-Candy SG, Farrant M, Gallo V. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat Neurosci. 2015;18(5):674-82
SLC4
Hentschke M, Wiemann M, Hentschke S, Kurth I, Seidenbecher T, Hermans-Borgmeyer I, Jentsch TJ, Gal A, Hübner CA (2006) Mice with a targeted disruption of the Cl-/HCO3- exchanger AE3 display a reduced seizure threshold. Molecular and Cellular Biology 26:182-91
Jacobs S, Ruusuvuori E. Sipilä S, Hapaanen A, Damkier HH, Kurth I, Hentschke M, Schweizer M, Rudhard Y, Laatinkainen L, Tyynelä J, Praetorius J, Voipio J, Hübner CA (2008) Targeted gene disruption of Slc4a10 results in reduced brain ventricle size and dampens neuronal excitability. Proceedings of the National Academy of Sciences U S A 105:311-6
Leviel F*, Hübner* CA, Morla L, Houillier P, El Moghrabi S, Brideau G, Hatim H, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, and Eladari D (2010) Identification of a novel amiloride-resistant, thiazide-sensitive NaCl reabsorption pathway in the distal nephron. Journal of Clinical Investigation 120(5):1627-35
Sinning A, Liebmann L, Kougioumtzes A, Westermann M, Bruehl C, Hübner CA (2011). Synaptic glutamate release is modulated by the Na+-driven Cl-/HCO₃- exchanger Slc4a8. Journal of Neuroscience 31(20):7300-11
Hilgen G, Huebner AK, Tanimoto N, Sothilingam V, Seide C, Garrido MG, Schmidt KF, Seeliger MW, Löwel S, Weiler R, Hübner CA*, Dedek K* (2012) Lack of the sodium-driven chloride bicarbonate exchanger NCBE impairs visual function in the mouse retina. PLoS One 7(10):e46155
Chambrey R, Kurth I, Peti-Peterdi J, Houillier P, Purkerson JM, Leviel F, Hentschke M, Zdebik AA, Schwartz GJ, Hübner CA, Eladari D (2013) Renal intercalated cells are rather energized by a proton than a sodium pump. Proceedings of the National Academy of Sciences U S A 110(19):7928-33
Mumtaz R, Trepiccione F, Hennings JC, Huebner AK, Serbin B, Picard N, Ullah AKMS, Păunescu TG, Capen DE, Lashhab RM, Mouro-Chanteloup I, Alper SL, Wagner CA, Cordat E, Brown D, Eladari D, Hübner CA (2017). Intercalated Cell Depletion and Vacuolar H+-ATPase Mistargeting in an Ae1 R607H Knockin Model. J Am Soc Nephrol 28(5):1507-1520
Axonopathies
Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape HC, Völkl H, Hübner CA, Jentsch TJ (2003) Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO Journal 22:5422-34
Hübner CA, Orth U, Senning A, Kohlschütter A, Korinthenberg R, Gal A (2005) 17 novel mutations in PLP1 causing Pelizaeus Merzbacher disease. Human Mutation 25:321-2
Hübner CA, Senning A, Zerres K, Gal A, Rudnik-Schöneborn S (2005) Mild Pelizaeus-Merzbacher disease caused by a point mutation affecting correct splicing of PLP1 mRNA. Neuroscience 132:687-701
Poët M, Kornak U, Schweizer M, Zdebik AA, Scheel O, Hoelter S, Wurst W, Schmitt A, Fuhrmann JC, Planells-Cases R, Mole SE, Hübner CA, Jentsch TJ (2006) Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proceedings of the National Academy of Sciences U S A 103:13854-9
Rust MB, Faulhaber J, Budack M, Pfeffer C, Maritzen T, Didié M, Beck FX, Boettger T, Schubert R, Ehmke H, Jentsch TJ, Hübner CA (2006) Neurogenic mechanisms contribute to hypertension in mice with disruption of the K-Cl-cotransporter KCC3. Circulation Research 98:549-56
Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, Rotthier A, Baets J, Senderek J, Topaloglu H, Farrel SA, Nürnberg P, De Jonghe P, Gal A, Kaether C, Timmerman V, Hübner CA (2009) Mutations in FAM134B, encoding a novel Golgi protein, cause severe sensory and autonomic neuropathy. Nature Genetics 41(11):1179-81
Beetz C, Koch N, Khundadze M, Zimmer G, Nietzsche S, Hertel N, Huebner AK, Mumtaz R, Schweizer M, Dirren E, Karle KN, Irintchev A, Alvarez V, Redies C, Westermann M, Kurth I, Deufel T, Kessels MM, Qualmann B, Hübner CA (2013) A spastic paraplegia mouse model reveals REEP1-dependent ER shaping. Journal of Clinical Investigation 123(10):4273-82
Khundadze M, Kollmann K, Koch N, Biskup C, Nietzsche S, Zimmer G, Hennings JC, Huebner AK, Symmank J, Jahic A, Ilina EI, Karle K, Schöls L, Kessels M, Braulke T, Qualmann B, Kurth I, Beetz C, Hübner CA (2013) A Hereditary Spastic Paraplegia Mouse Model Supports a Role of ZFYVE26/SPASTIZIN for the Endolysosomal System. PLoS Genetics 9(12):e1003988
Hübner CA, Kurth I. Membrane-shaping disorders: a common pathway in axon degeneration. Brain. 2014;137(Pt 12):3109-21
Leipold E, Liebmann L, Korenke GC, Heinrich T, Giesselmann S, Baets J, Ebbinghaus M, Goral RO, Stödberg T, Hennings JC, Bergmann M, Altmüller J, Thiele H, Wetzel A, Nürnberg P, Timmerman V, De Jonghe P, Blum R, Schaible HG, Weis J, Heinemann SH, Hübner CA, Kurth I (2013) A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nature Genetics 45(11):1399-404
Koehler K, Malik M, Mahmood S, Gießelmann S, Beetz C, Hennings JC, Huebner AK, Grahn A, Reunert J, Nürnberg G, Thiele H, Altmüller J, Nürnberg P, Mumtaz R, Babovic-Vuksanovic D, Basel-Vanagaite L, Borck G, Brämswig J, Mühlenberg R, Sarda P, Sikiric A, Anyane-Yeboa K, Zeharia A, Ahmad A, Coubes C, Wada Y, Marquardt T, Vanderschaeghe D, Van Schaftingen E, Kurth I, Huebner A, Hübner CA (2013) Mutations in GMPPA cause a glycosylation disorder characterized by intellectual disability and autonomic dysfunction. American Journal of Human Genetics 93(4):727-34
Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M, Katona I, Qualmann B, Weis J, Reggiori F, Kurth I*, Hübner CA*, Dikic I*. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522(7556):354-8 *equal contribution
Varga RE, Khundadze M, Damme M, Nietzsche S, Hoffmann B, Stauber T, Koch N, Hennings JC, Franzka P, Huebner AK, Kessels MM, Biskup C, Jentsch TJ, Qualmann B, Braulke T, Kurth I, Beetz C, Hübner CA. In Vivo Evidence for Lysosome Depletion and Impaired Autophagic Clearance in Hereditary Spastic Paraplegia Type SPG11. PLoS Genet. 2015;11(8):e1005454